We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
General description of the gene and encoded protein(s) using information from HGNC and Ensembl, as well as predictions made as well as predictions made by the Human Protein Atlas project.
All transcripts of all genes have been analyzed regarding the location(s) of corresponding protein based on prediction methods for signal peptides and transmembrane regions.
Genes with at least one transcript predicted to encode a secreted protein, according to prediction methods or to UniProt location data, have been further annotated and classified with the aim to determine if the corresponding protein(s) are secreted or actually retained in intracellular locations or membrane-attached.
Remaining genes, with no transcript predicted to encode a secreted protein, will be assigned the prediction-based location(s).
The annotated location overrules the predicted location, so that a gene encoding a predicted secreted protein that has been annotated as intracellular will have intracellular as the final location.
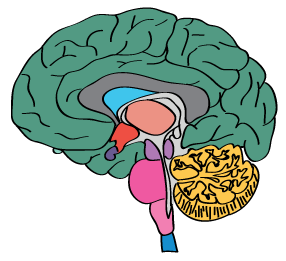
The RNA specificity category is based on mRNA expression levels in the analyzed samples based on a combination of data from HPA, GTEX and FANTOM5. The categories include: tissue enriched, group enriched, tissue enhanced, low tissue specificity and not detected. The Tissue classification is based on 37 human tissue types, where brain is represented by the highest expression value among the human brain regions (including spinal cord and corpus callosum that is excluded from the regional classification based on 10 main brain regions). The combination of tissue and regional classification can be found here
The regional specificity category is based on mRNA expression levels in the analyzed brain samples, grouped into 10 main brain regions and calculated for the three different species. The human brain expression profile is based on a combination of data from GTEX and FANTOM5. The specificity categories include: regionally enriched, group enriched, regionally enhanced, low regional specificity and not detected. The classification rules are the same used for the tissue specificity category.
Group enriched (basal ganglia, hypothalamus, pons and medulla)
Group enriched (basal ganglia, pons and medulla)
Low region specificity
Regional distributioni
The regional distribution category is based on mRNA expression detected above cut off or not in the analyzed brain samples, grouped into 10 main brain regions and calculated for the three different species. The human brain expression is based on a combination of data from GTEX and FANTOM5. The distribution categories include: detected in all, detected in many, detected in some, detected in single and not detected. The classification rules are the same used for the tissue distribution category.
A summary of the overall protein localization across the analyzed brain tissue. The summary is based on manual annotation.
Mouse brain profiles includes a more extensive summary of the protein location while summaries with only human protein profile are more limited and are based on imported data/images from the Tissue Atlas stainings that has undergone an additional brain-centric curation step.
Immunoreactivity in cholinergic neuronal fibers throughout the brain, highly in basal lateral amygdala and interpeduncular nucleus fibers. Strong expression in soma of motor neurons (V), facial nucleus, hypoglossal.
Consensus normalized expression (NX) levels were created for the 10 brain regions by combining the data from two transcriptomics datasets (GTEx and FANTOM5). Color coding is based on brain region and the bar shows the highest expression among the subregions included. To access sample data, click on region name or bar.
Read more about how the datasets were combined into consensus normalized expression levels in Assays & Annotation.
GTEx Human brain RNA-Seq dataseti
GTEx dataset RNA-seq tissue data generated by the Genotype-Tissue Expression (GTEx) project is reported as mean pTPM (protein-coding transcripts per million), corresponding to mean values of the different individual samples for respective subregion. Highest expression among the subregions represents the brain region. To access sample data, click on region name or bar.
FANTOM5 dataset Tissue data for RNA expression obtained through Cap Analysis of Gene Expression (CAGE) generated by the FANTOM5 project are reported as Scaled Tags Per Million. To access sample data, click on region name or bar.
HPA Pig dataset HPA RNA-seq tissue data is reported as mean NX (normalized expression) for the 10 brain regions. The NX calculation is based on pTPM (protein-coding transcripts per million) of the individual samples, avaliable at the detaile pages (reached when clicking a bar or regional name). To access sample data, click on region name or bar.
The pig brain transcriptomics project is a collaborative project between human protein atlas and the Lars Bolund institute of regenerative Medicine (Dr. Yonglun Luo), BGI-Qingdao, China.
HPA Mouse dataset HPA RNA-seq tissue data is reported as mean NX (normalized expression) for the 10 brain regions. The NX calculation is based on pTPM (protein-coding transcripts per million) of the individual samples, avaliable at the detaile pages (reached when clicking a bar or regional name). To access sample data, click on region name or bar.
Mouse brain protein profiles are based in serial sections of the adult mouse brain analyzed by immunofuorescence. The staining intensity and regional distribution is manually summarized.
Immunoreactivity in cholinergic neuronal fibers throughout the brain, highly in basal lateral amygdala and interpeduncular nucleus fibers. Strong expression in soma of motor neurons (V), facial nucleus, hypoglossal.
Cellular location
Axon and synapse in neurons.
Protein profile reliability
Supported based on CAB079011
HPA Mouse staining intesityi
The score is based on the relative fluorescence, defined as intensity of the annotated region relative to the intensity of the region with highest intensity. The digitalized images are processed (axel-adjusted and tissue edges defined) and regions of interest (ROIs) are then marked. These ROIs are then used for image analysis and the relative fluorescence intensity is listed for each region. For more data and regional information click on the bar or the regional name.
 The Human Protein Atlas project is funded
The Human Protein Atlas project is funded



 MENU
MENU